Development of Cancer Prognostic Signature Based on Pan-Cancer Proteomics
Abstract
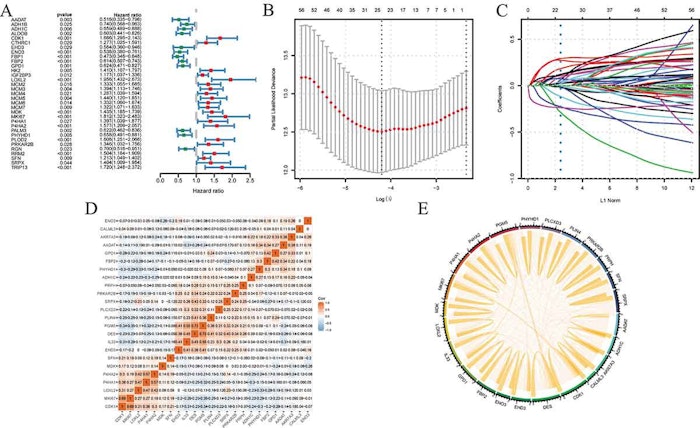
Utilizing genomic data to predict cancer prognosis was insufficient. Proteomics can improve our understanding of the etiology and progression of cancer and improve the assessment of cancer prognosis. And the Clinical Proteomic Tumor Analysis Consortium (CPTAC) has generated extensive proteomics data of the vast majority of tumors. Based on CPTAC, we can perform a proteomic pan-carcinoma analysis. We collected the proteomics data and clinical features of cancer patients from CPTAC. Then, we screened 69 differentially expressed proteins (DEPs) with R software in five cancers: hepatocellular carcinoma (HCC), children's brain tumor tissue consortium (CBTTC), clear cell renal cell carcinoma (CCRC), lung adenocarcinoma (LUAD) and uterine corpus endometrial carcinoma (UCEC). GO and KEGG analysis were performed to clarify the function of these proteins. We also identified their interactions. The DEPs-based prognostic model for predicting over survival was identified by least absolute shrinkage and selection operator (LASSO)-Cox regression model in training cohort. Then, we used the time-dependent receiver operating characteristics analysis to evaluate the ability of the prognostic model to predict overall survival and validated it in validation cohort. The results showed that the DEPs-based prognostic model could accurately and effectively predict the survival rate of most cancers.
Highlights:
- 69 differentially expressed proteins (DEPs) were identified.
- The DEPs formed an interaction network across five cancers.
- The 24 DEPs could accurately predict the OS in multiple cancers.