Novel findings with reassessment of exome data: implications for validation testing and interpretation of genomic data
Abstract
Purpose
The objective of this study was to assess the ability of our laboratory’s exome-sequencing test to detect known and novel sequence variants and identify the critical factors influencing the interpretation of a clinical exome test.
Methods
We developed a two-tiered validation strategy: (i) a method-based approach that assessed the ability of our exome test to detect known variants using a reference HapMap sample, and (ii) an interpretation-based approach that assessed our relative ability to identify and interpret disease-causing variants, by analyzing and comparing the results of 19 randomly selected patients previously tested by external laboratories.
Results
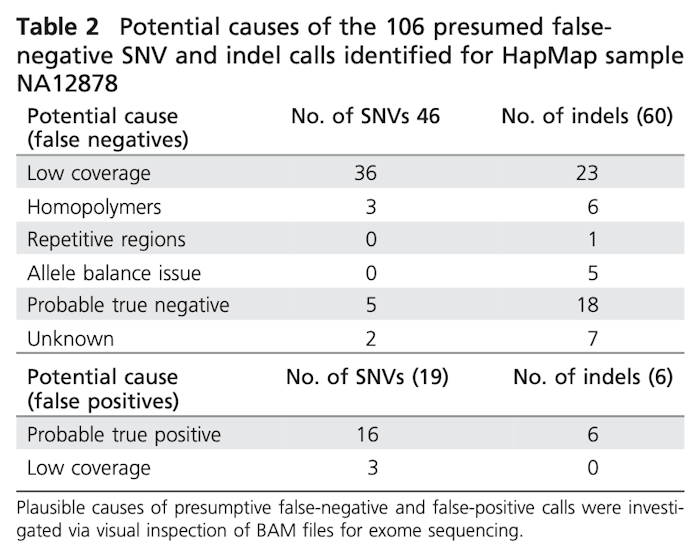
We demonstrate that this approach is reproducible with >99% analytical sensitivity and specificity for single-nucleotide variants and indels <10 bp. Our findings were concordant with the reference laboratories in 84% of cases. A new molecular diagnosis was applied to three cases, including discovery of two novel candidate genes.
Conclusion
We provide an assessment of critical areas that influence interpretation of an exome test, including comprehensive phenotype capture, assessment of clinical overlap, availability of parental data, and the addressing of limitations in database updates. These results can be used to inform improvements in phenotype-driven interpretation of medical exomes in clinical and research settings.