Comprehensive Molecular Profiling of Pediatric Meningioma
Email Principal Investigator

Kathleen M. Schieffer
CBTN Data
About this
Project
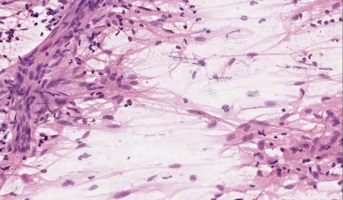
Meningiomas are the most common tumors occurring in the brain and spine. Pediatric meningiomas, however, are extremely rare tumors and are often of the most aggressive form. Furthermore, a majority of these tumors are associated with a well-described genetic disorder, neurofibromatosis type 2 (NF2). The mechanisms involved in NF2 driven meningiomas is well described, but there are many other forms of meningioma whose genetic drivers are not yet well understood, including gene fusions. A fusion gene is a gene made up of two previously independent genes. An example of such a fusion is a YAP1-FAM118B fusion recently located in a pediatric meningioma patient enrolled in cancer protocol at Nationwide Children’s Hospital (NCH). Using invaluable data from the Pediatric Brain Tumor Atlas, researchers will compare YAP1 fusion meningioma to pediatric meningioma driven by other genetic drivers, including NF2, AKT1, TRAF7, and SMO in an effort to identify opportunities for new therapies.
Ask The
Scientists

What are the goals of this project?
Researchers seek to compare and contrast rare pediatric meningioma mutations in an effort to propel research into new treatments.
What is the impact of this project?
A comprehensive look at poorly misunderstood pediatric meningiomas will pave the way for more accurate diagnostics and future therapies, particularly in patients whose tumors are not able to be fully removed in surgery. The data generated will provide a deeper characterization of this tumor type and the data will be shared with the community.
Why is the CBTN request important to this project?
Data on pediatric meningiomas is rare. The CBTN has a cohort of 11 pediatric meningioma tumors and of these cases, one patient has the same fusion event (YAP1- FAM118B) as a patient recently identified in another study. This comparative data is crucial for the execution of this project.
Specimen Data
The Children's Brain Tumor Network contributed to this project by providing access to the Pediatric Brain Tumor Atlas.
Explore the data in these informatics portals

Meet The